Deck 19: Cancer and Regulation of the Cell Cycle
Full screen (f)
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
Question
The table in this problem summarizes some of the data that have been collected on BRCA1 mutations in families with a high incidence of both early-onset breast cancer and ovarian cancer.
Predisposing Mutations in BRCA 1
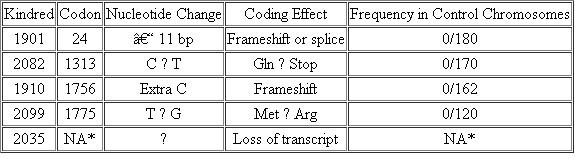 Source : 1994. Science 266: 66-71. © AAAS.
Source : 1994. Science 266: 66-71. © AAAS.
*NA indicates not applicable, as the regulatory mutation is inferred, and the position has not been identified.
(a) Note the coding effect of the mutation found in kindred group 2082. This results from a single base-pair substitution. Draw the normal double-stranded DNA sequence for this codon (with the 5? and 3? ends labeled), and show the sequence of events that generated this mutation, assuming that it resulted from an uncorrected mismatch event during DNA replication.
(b) Examine the types of mutations that are listed in the table and determine if the BRCA1 gene is likely to be a tumor-suppressor gene or an oncogene.
(c) Although the mutations listed in the table are clearly deleterious and cause breast cancer in women at very young ages, each of the kindred groups had at least one woman who carried the mutation but lived until age 80 without developing cancer. Name at least two different mechanisms (or variables) that could underlie variation in the expression of a mutant phenotype and propose an explanation for the incomplete penetrance of this mutation. How do these mechanisms or variables relate to this explanation?
Predisposing Mutations in BRCA 1
Source : 1994. Science 266: 66-71. © AAAS.*NA indicates not applicable, as the regulatory mutation is inferred, and the position has not been identified.
(a) Note the coding effect of the mutation found in kindred group 2082. This results from a single base-pair substitution. Draw the normal double-stranded DNA sequence for this codon (with the 5? and 3? ends labeled), and show the sequence of events that generated this mutation, assuming that it resulted from an uncorrected mismatch event during DNA replication.
(b) Examine the types of mutations that are listed in the table and determine if the BRCA1 gene is likely to be a tumor-suppressor gene or an oncogene.
(c) Although the mutations listed in the table are clearly deleterious and cause breast cancer in women at very young ages, each of the kindred groups had at least one woman who carried the mutation but lived until age 80 without developing cancer. Name at least two different mechanisms (or variables) that could underlie variation in the expression of a mutant phenotype and propose an explanation for the incomplete penetrance of this mutation. How do these mechanisms or variables relate to this explanation?
Question
The following table shows neutral polymorphisms found in control families (those with no increased frequency of breast and ovarian cancer).
Neutral Polymorphisms in BRCA 1
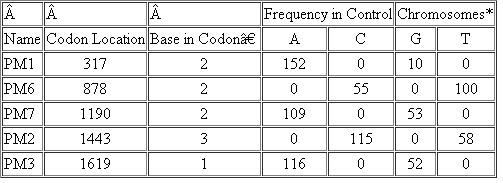 *The number of chromosomes with a particular base at the indicated polymorphic site (A, C, G, or T) is shown.
*The number of chromosomes with a particular base at the indicated polymorphic site (A, C, G, or T) is shown.
▪Position 1, 2, or 3 of the codon.
Examine the data in the table and answer the following questions:
(a) What is meant by a neutral polymorphism?
(b) What is the significance of this table in the context of examining a family or population for BRCA1 mutations that predispose an individual to cancer?
(c) Is the PM2 polymorphism likely to result in a neutral mis-sense mutation or a silent mutation?
(d) Answer part (c) for the PM3 polymorphism.
Neutral Polymorphisms in BRCA 1
*The number of chromosomes with a particular base at the indicated polymorphic site (A, C, G, or T) is shown.▪Position 1, 2, or 3 of the codon.
Examine the data in the table and answer the following questions:
(a) What is meant by a neutral polymorphism?
(b) What is the significance of this table in the context of examining a family or population for BRCA1 mutations that predispose an individual to cancer?
(c) Is the PM2 polymorphism likely to result in a neutral mis-sense mutation or a silent mutation?
(d) Answer part (c) for the PM3 polymorphism.
Question
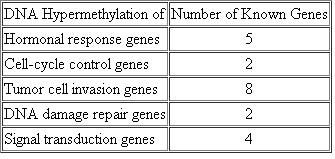
Prostate cancer is a major cause of cancer-related deaths among men. Epigenetic changes that regulate gene expression are involved in both the initiation and progression of such cancers. Following is a table that lists the number of genes known to be hypermethylated in prostate cancer cells (modified from Long-Cheng, L. et al., 2005. J. Natl. Cancer Inst. 97: 103-115). For each category of genes, speculate on the mechanism(s) by which cancer initiation or progression might be influenced by hypermethylation.

Unlock Deck
Sign up to unlock the cards in this deck!
Unlock Deck
Unlock Deck
1/35
Play
Full screen (f)
Deck 19: Cancer and Regulation of the Cell Cycle
1
A middle-aged woman taking the breast cancer drug Tamoxifen for ten years became concerned when she saw a news report with disturbing information. In some women, the drug made their cancer more aggressive and more likely to spread. Other women with breast cancer, the report stated, do not respond to Tamoxifen at all, and 30 to 40 percent of women who take the drug eventually become resistant to chemotherapy. The woman contacted her oncologist to ask some questions:
How can some people react one way to a cancer treatment and others react a different way?
How can some people react one way to a cancer treatment and others react a different way?
Every person is genetically unique. For this reason, the genetic makeup of cancers cells in one individual is different from every other individual. In addition, cancer is the result of a plethora of mutations. Even among the same types of cancer, the pathway to tumor development will vary widely between individuals.
▪specific chemotherapeutic agent may only target a specific receptor or protein expressed in a small subset of patients. A prime example is the breast cancer drug Herceptin®. It targets and blocks a specific receptor (human epidermal growth factor receptor 2 or HER2) in breast cancer cells that stimulates uncontrolled cell proliferation. However, not all breast cancer patients express the HER2 receptor and Herceptin® is not effective in these patients.
▪specific chemotherapeutic agent may only target a specific receptor or protein expressed in a small subset of patients. A prime example is the breast cancer drug Herceptin®. It targets and blocks a specific receptor (human epidermal growth factor receptor 2 or HER2) in breast cancer cells that stimulates uncontrolled cell proliferation. However, not all breast cancer patients express the HER2 receptor and Herceptin® is not effective in these patients.
2
In this chapter, we focused on cancer as a genetic disease, with an emphasis on the relationship between cancer, the cell cycle, and DNA damage, as well as on the multiple steps that lead to cancer. At the same time, we found many opportunities to consider the methods and reasoning by which much of this information was acquired. From the explanations given in the chapter,
(a) How do we know that malignant tumors arise from a single cell that contains mutations?
(b) How do we know that cancer development requires more than one mutation?
(c) How do we know that cancer cells contain defects in DNA repair?
(a) How do we know that malignant tumors arise from a single cell that contains mutations?
(b) How do we know that cancer development requires more than one mutation?
(c) How do we know that cancer cells contain defects in DNA repair?
(a)Several pieces of evidence support the clonal origin of primary and secondary tumors. Regardless of the type of cancer, each cancer cell in an individuals' body contains the same genetic aberration(s) which caused the cancer. This suggests that tumors (consisting of billions of cells) originated from a single mutated cell.
Also, all cancer cells in and individual female contain the same inactivated X-chromosome. X-chromosome inactivation is the process by which one of the two copies of the X chromosome in females is deactivated early in embryonic development to prevent females from having twice the number of gene products. This occurs at random and the cells of females contain a mixture of inactive maternal and paternal X-chromosomes.
(b)The increased risk of cancer as people age suggests that multiple mutations must occur before cancer develops. Also, carcinogenic exposure provides clues that single mutations don't give rise to tumors. For example, in skin cancer, it takes many years of ultraviolet (UV) light exposure before the development of tumors, indicating an accumulation of multiple mutations.
Lastly, spontaneous mutations in humans occur at a rate of approximately 10 -6 mutations per gene, per cell division. At this rate, single mutations anywhere in the body would be abundant (approximately 10 10 per gene) over the course of a lifetime. If tumors arose from single mutations, this many mutations would undoubtedly result in cancer in every person, but the approximate rate of cancer in humans is 1 in 3. In addition, mutated cells tend to replicate more frequently, and knowing the clonal nature of cancer, these frequently replicating cells have a higher chance of acquiring more mutations through each round of replication.
(c)
The mutator phenotype theorizes that mutations in early tumors are significantly more prevalent than mutations in normal somatic cells. This suggests that with the low error rate of deoxyribonucleic acid (DNA) replication in normal cells and large number of mutations (can be in the thousands) in cancer cells, a defect in the repair mechanism of DNA must be one cause.
Molecular characterization has also provided evidence that defective DNA repair mechanisms give rise to cancers. Different DNA repair mechanisms correct damage at the DNA sequence and chromosome level through direct base or nucleotide repair to cell cycle arrest and apoptosis. A classic example is analysis of cells from individuals diagnosed with xeroderma pigmentosum (XP), a disorder exhibiting heightened sensitivity to certain carcinogens, particularly UV light. Cells in XP individuals were found to be defective in nucleotide excision repair, a mechanism that can repair thymine dimers caused by UV light exposure. Another example is the RAD group of genes, which encode several different DNA repair proteins, which have been implicated in a variety of cancers.
Also, all cancer cells in and individual female contain the same inactivated X-chromosome. X-chromosome inactivation is the process by which one of the two copies of the X chromosome in females is deactivated early in embryonic development to prevent females from having twice the number of gene products. This occurs at random and the cells of females contain a mixture of inactive maternal and paternal X-chromosomes.
(b)The increased risk of cancer as people age suggests that multiple mutations must occur before cancer develops. Also, carcinogenic exposure provides clues that single mutations don't give rise to tumors. For example, in skin cancer, it takes many years of ultraviolet (UV) light exposure before the development of tumors, indicating an accumulation of multiple mutations.
Lastly, spontaneous mutations in humans occur at a rate of approximately 10 -6 mutations per gene, per cell division. At this rate, single mutations anywhere in the body would be abundant (approximately 10 10 per gene) over the course of a lifetime. If tumors arose from single mutations, this many mutations would undoubtedly result in cancer in every person, but the approximate rate of cancer in humans is 1 in 3. In addition, mutated cells tend to replicate more frequently, and knowing the clonal nature of cancer, these frequently replicating cells have a higher chance of acquiring more mutations through each round of replication.
(c)
The mutator phenotype theorizes that mutations in early tumors are significantly more prevalent than mutations in normal somatic cells. This suggests that with the low error rate of deoxyribonucleic acid (DNA) replication in normal cells and large number of mutations (can be in the thousands) in cancer cells, a defect in the repair mechanism of DNA must be one cause.
Molecular characterization has also provided evidence that defective DNA repair mechanisms give rise to cancers. Different DNA repair mechanisms correct damage at the DNA sequence and chromosome level through direct base or nucleotide repair to cell cycle arrest and apoptosis. A classic example is analysis of cells from individuals diagnosed with xeroderma pigmentosum (XP), a disorder exhibiting heightened sensitivity to certain carcinogens, particularly UV light. Cells in XP individuals were found to be defective in nucleotide excision repair, a mechanism that can repair thymine dimers caused by UV light exposure. Another example is the RAD group of genes, which encode several different DNA repair proteins, which have been implicated in a variety of cancers.
3
A middle-aged woman taking the breast cancer drug Tamoxifen for ten years became concerned when she saw a news report with disturbing information. In some women, the drug made their cancer more aggressive and more likely to spread. Other women with breast cancer, the report stated, do not respond to Tamoxifen at all, and 30 to 40 percent of women who take the drug eventually become resistant to chemotherapy. The woman contacted her oncologist to ask some questions:
Why do most cancers eventually become resistant to a specific chemotherapeutic drug?
Why do most cancers eventually become resistant to a specific chemotherapeutic drug?
Tumors contain a mixture of cancer cells that are drug-sensitive or drug-resistant. Chemotherapy wipes out the drug-sensitive cells while the drug-resistant cells remain and continue to proliferate. Over time, the tumor becomes largely composed of drug-resistant cells and chemotherapy drugs cease to be effective.
Research has elucidated several mechanisms by which cancer cells become multidrug resistant. One such method is molecular pumps embedded in the cell membrane of cancer cells that effectively eject chemotherapeutic drugs from the cell.
Epigenetic effects also account for drug resistance. Epigenetic effects are changes in gene expression or activity, not by underlying changes in deoxyribonucleic acid (DNA), but through other mechanisms that alter gene expression activity. In cancer cells, exposure to a chemotherapeutic drug can initiate a cascade of events that alters gene expression activity. The altered gene expression can activate certain proteins and processes that confer resistance to chemotherapeutic drugs.
Research has elucidated several mechanisms by which cancer cells become multidrug resistant. One such method is molecular pumps embedded in the cell membrane of cancer cells that effectively eject chemotherapeutic drugs from the cell.
Epigenetic effects also account for drug resistance. Epigenetic effects are changes in gene expression or activity, not by underlying changes in deoxyribonucleic acid (DNA), but through other mechanisms that alter gene expression activity. In cancer cells, exposure to a chemotherapeutic drug can initiate a cascade of events that alters gene expression activity. The altered gene expression can activate certain proteins and processes that confer resistance to chemotherapeutic drugs.
4
Review the Chapter Concepts list. These concepts relate to the multiple ways in which genetic alterations lead to the development of cancers. The sixth concept states that epigenetic effects including DNA methylation and histone modifications contribute to the genetic alterations leading to cancer. Write a short essay describing how epigenetic changes in cancer cells contribute to the development of cancers.
▪Cancer is characterized by genetic defects in fundamental aspects of cellular function, including DNA repair, chromatin modification, cell-cycle regulation, apoptosis, and signal transduction.
▪Most cancer-causing mutations occur in somatic cells; only about 5 percent of cancers have a hereditary component.
▪Mutations in cancer-related genes lead to abnormal proliferation and loss of control over how cells spread and invade surrounding tissues.
▪The development of cancer is a multistep process requiring mutations in genes controlling many aspects of cell proliferation and metastasis.
▪Cancer cells show high levels of genomic instability, leading to the accumulation of multiple mutations, some in cancer-related genes.
▪Epigenetic effects such as DNA methylation and histone modifications play significant roles in the development of cancers.
▪Mutations in proto-oncogenes and tumor-suppressor genes contribute to the development of cancers.
▪Cancer-causing viruses introduce oncogenes into infected cells and stimulate cell proliferation.
▪Environmental agents contribute to cancer by damaging DNA.
▪Cancer is characterized by genetic defects in fundamental aspects of cellular function, including DNA repair, chromatin modification, cell-cycle regulation, apoptosis, and signal transduction.
▪Most cancer-causing mutations occur in somatic cells; only about 5 percent of cancers have a hereditary component.
▪Mutations in cancer-related genes lead to abnormal proliferation and loss of control over how cells spread and invade surrounding tissues.
▪The development of cancer is a multistep process requiring mutations in genes controlling many aspects of cell proliferation and metastasis.
▪Cancer cells show high levels of genomic instability, leading to the accumulation of multiple mutations, some in cancer-related genes.
▪Epigenetic effects such as DNA methylation and histone modifications play significant roles in the development of cancers.
▪Mutations in proto-oncogenes and tumor-suppressor genes contribute to the development of cancers.
▪Cancer-causing viruses introduce oncogenes into infected cells and stimulate cell proliferation.
▪Environmental agents contribute to cancer by damaging DNA.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
5
A middle-aged woman taking the breast cancer drug Tamoxifen for ten years became concerned when she saw a news report with disturbing information. In some women, the drug made their cancer more aggressive and more likely to spread. Other women with breast cancer, the report stated, do not respond to Tamoxifen at all, and 30 to 40 percent of women who take the drug eventually become resistant to chemotherapy. The woman contacted her oncologist to ask some questions:
Why does it seem that some drugs are thought to be safe one day and declared unsafe the next day?
Why does it seem that some drugs are thought to be safe one day and declared unsafe the next day?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
6
Where are the major regulatory points in the cell cycle?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
7
List the functions of kinases and cyclins, and describe how they interact to cause cells to move through the cell cycle.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
8
How does pRB function to keep cells at the G1 checkpoint?
(b) How do cells get past the G1 checkpoint to move into S phase?
(b) How do cells get past the G1 checkpoint to move into S phase?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
9
What is the difference between saying that cancer is inherited and saying that the predisposition to cancer is inherited?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
10
As a genetic counselor, you are asked to assess the risk for a couple with a family history of retinoblastoma who are thinking about having children. Both the husband and wife are phenotypically normal, but the husband has a sister with familial retinoblastoma in both eyes. What is the probability that this couple will have a child with retinoblastoma? Are there any tests that you could recommend to help in this assessment?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
11
What is apoptosis, and under what circumstances do cells undergo this process?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
12
Define tumor-suppressor genes. Why is a mutated single copy of a tumor-suppressor gene expected to behave as a recessive gene?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
13
A genetic variant of the retinoblastoma protein, called PSM-RB (phosphorylation site mutated RB), is not able to be phos-phorylated by the action of CDK4/cyclin D1 complex. Explain why PSM-RB is said to have a constitutive growth-suppressing action on the cell cycle.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
14
Part of the Ras protein is associated with the plasma membrane, and part extends into the cytoplasm. How does the Ras protein transmit a signal from outside the cell into the cytoplasm? What happens in cases where the ras gene is mutated?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
15
If a cell suffers damage to its DNA while in S phase, how can this damage be repaired before the cell enters mitosis?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
16
Distinguish between oncogenes and proto-oncogenes. In what ways can proto-oncogenes be converted to oncogenes?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
17
Of the two classes of genes associated with cancer, tumor-suppressor genes and oncogenes, mutations in which group can be considered gain-of-function mutations? In which group are the loss-of-function mutations? Explain.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
18
How do translocations such as the Philadelphia chromosome contribute to cancer?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
19
Explain why many oncogenic viruses contain genes whose products interact with tumor-suppressor proteins.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
20
DNA sequencing has provided data to indicate that cancer cells may contain tens of thousands of somatic mutations, only some of which confer a growth advantage to a cancer cell. How do scientists describe and categorize these recently discovered populations of mutations in cancer cells?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
21
How do normal cells protect themselves from accumulating mutations in genes that could lead to cancer? How do cancer cells differ from normal cells in these processes?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
22
Describe the difference between an acute transforming virus and a virus that does not cause tumors.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
23
Epigenetics is a relatively new area of genetics with a focus on phenomena that affect gene expression but do not affect DNA sequence. Epigenetic effects are quasi-stable and may be passed to progeny somatic or germ-line cells. What are known causes of epigenetic effects, and how do they relate to cancer?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
24
Radiotherapy (treatment with ionizing radiation) is one of the most effective current cancer treatments. It works by damaging DNA and other cellular components. In which ways could radiotherapy control or cure cancer, and why does radiotherapy often have significant side effects?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
25
Genetic tests that detect mutations in the BRCA1 and BRCA2 oncogenes are widely available. These tests reveal a number of mutations in these genes-mutations that have been linked to familial breast cancer. Assume that a young woman in a suspected breast cancer family takes the BRCA1 and BRCA2 genetic tests and receives negative results. That is, she does not test positive for the mutant alleles of BRCA1 or BRCA2. Can she consider herself free of risk for breast cancer?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
26
Explain the apparent paradox that both hypermethylation and hypomethylation of DNA are often found in the same cancer cell.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
27
As part of a cancer research project, you have discovered a gene that is mutated in many metastatic tumors. After determining the DNA sequence of this gene, you compare the sequence with those of other genes in the human genome sequence database. Your gene appears to code for an amino acid sequence that resembles sequences found in some serine proteases. Conjecture how your new gene might contribute to the development of highly invasive cancers.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
28
Describe the steps by which the p53 gene responds to DNA damage and/or cellular stress to promote cell-cycle arrest and apoptosis. Given that p53 is a recessive gene and is not located on the X chromosome, why would people who inherit just one mutant copy of a recessive tumor-suppressor gene be at higher risk of developing cancer than those without the recessive gene?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
29
Mutations in tumor-suppressor genes are associated with many types of cancers. In addition, epigenetic changes (such as DNA methylation) of tumor-suppressor genes are also associated with tumorigenesis (Otani et al., 2013. Expert Rev Mol Diagn 13: 445-455).
(a) How might hypermethylation of the p53 gene promoter influence tumorigenesis?
(b) Knowing that tumors release free DNA into certain surrounding body fluids through necrosis and apoptosis (Kloten et al., 2013. Breast Cancer Res. 15(1): R4), outline an experimental protocol for using human blood as a biomarker for cancer and as a method for monitoring the progression of cancer in an individual.
(a) How might hypermethylation of the p53 gene promoter influence tumorigenesis?
(b) Knowing that tumors release free DNA into certain surrounding body fluids through necrosis and apoptosis (Kloten et al., 2013. Breast Cancer Res. 15(1): R4), outline an experimental protocol for using human blood as a biomarker for cancer and as a method for monitoring the progression of cancer in an individual.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
30
Vanderbilt University Medical Center maintains a Web site (http://bioinfo.mc.vanderbilt.edu/TSGene/) that contains descriptions of tumor-suppressor genes, including 637 protein-coding genes and 79 noncoding segments of DNA. How can noncoding segments of DNA function or produce products that function as tumor suppressors?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
31
A study by Bose and colleagues (1998. Blood 92: 3362-3367) and a previous study by Biernaux and others (1996. Bone Marrow Transplant 17: (Suppl. 3) S45-S47) showed that BCR-ABL fusion gene transcripts can be detected in 25 to 30 percent of healthy adults who do not develop chronic myelogenous leukemia (CML). Explain how these individuals can carry a fusion gene that is transcriptionally active and yet do not develop CML.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
32
Those who inherit a mutant allele of the RB1 gene are at risk for developing a bone cancer called osteosarcoma. You suspect that in these cases, osteosarcoma requires a mutation in the second RB1 allele, and you have cultured some osteosarcoma cells and obtained a cDNA clone of a normal human RB1 gene. A colleague sends you a research paper revealing that a strain of cancer-prone mice develop malignant tumors when injected with osteosarcoma cells, and you obtain these mice. Using these three resources, what experiments would you perform to determine (a) whether osteosarcoma cells carry two RB1 mutations, (b) whether osteosarcoma cells produce any pRB protein, and (c) if the addition of a normal RB1 gene will change the cancer-causing potential of osteosarcoma cells?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
33
The table in this problem summarizes some of the data that have been collected on BRCA1 mutations in families with a high incidence of both early-onset breast cancer and ovarian cancer.
Predisposing Mutations in BRCA 1
Source : 1994. Science 266: 66-71. © AAAS.
*NA indicates not applicable, as the regulatory mutation is inferred, and the position has not been identified.
(a) Note the coding effect of the mutation found in kindred group 2082. This results from a single base-pair substitution. Draw the normal double-stranded DNA sequence for this codon (with the 5? and 3? ends labeled), and show the sequence of events that generated this mutation, assuming that it resulted from an uncorrected mismatch event during DNA replication.
(b) Examine the types of mutations that are listed in the table and determine if the BRCA1 gene is likely to be a tumor-suppressor gene or an oncogene.
(c) Although the mutations listed in the table are clearly deleterious and cause breast cancer in women at very young ages, each of the kindred groups had at least one woman who carried the mutation but lived until age 80 without developing cancer. Name at least two different mechanisms (or variables) that could underlie variation in the expression of a mutant phenotype and propose an explanation for the incomplete penetrance of this mutation. How do these mechanisms or variables relate to this explanation?
Predisposing Mutations in BRCA 1
Source : 1994. Science 266: 66-71. © AAAS.*NA indicates not applicable, as the regulatory mutation is inferred, and the position has not been identified.
(a) Note the coding effect of the mutation found in kindred group 2082. This results from a single base-pair substitution. Draw the normal double-stranded DNA sequence for this codon (with the 5? and 3? ends labeled), and show the sequence of events that generated this mutation, assuming that it resulted from an uncorrected mismatch event during DNA replication.
(b) Examine the types of mutations that are listed in the table and determine if the BRCA1 gene is likely to be a tumor-suppressor gene or an oncogene.
(c) Although the mutations listed in the table are clearly deleterious and cause breast cancer in women at very young ages, each of the kindred groups had at least one woman who carried the mutation but lived until age 80 without developing cancer. Name at least two different mechanisms (or variables) that could underlie variation in the expression of a mutant phenotype and propose an explanation for the incomplete penetrance of this mutation. How do these mechanisms or variables relate to this explanation?
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
34
The following table shows neutral polymorphisms found in control families (those with no increased frequency of breast and ovarian cancer).
Neutral Polymorphisms in BRCA 1
*The number of chromosomes with a particular base at the indicated polymorphic site (A, C, G, or T) is shown.
▪Position 1, 2, or 3 of the codon.
Examine the data in the table and answer the following questions:
(a) What is meant by a neutral polymorphism?
(b) What is the significance of this table in the context of examining a family or population for BRCA1 mutations that predispose an individual to cancer?
(c) Is the PM2 polymorphism likely to result in a neutral mis-sense mutation or a silent mutation?
(d) Answer part (c) for the PM3 polymorphism.
Neutral Polymorphisms in BRCA 1
*The number of chromosomes with a particular base at the indicated polymorphic site (A, C, G, or T) is shown.▪Position 1, 2, or 3 of the codon.
Examine the data in the table and answer the following questions:
(a) What is meant by a neutral polymorphism?
(b) What is the significance of this table in the context of examining a family or population for BRCA1 mutations that predispose an individual to cancer?
(c) Is the PM2 polymorphism likely to result in a neutral mis-sense mutation or a silent mutation?
(d) Answer part (c) for the PM3 polymorphism.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
35
Prostate cancer is a major cause of cancer-related deaths among men. Epigenetic changes that regulate gene expression are involved in both the initiation and progression of such cancers. Following is a table that lists the number of genes known to be hypermethylated in prostate cancer cells (modified from Long-Cheng, L. et al., 2005. J. Natl. Cancer Inst. 97: 103-115). For each category of genes, speculate on the mechanism(s) by which cancer initiation or progression might be influenced by hypermethylation.
Unlock Deck
Unlock for access to all 35 flashcards in this deck.
Unlock Deck
k this deck
Unlock Deck
Unlock for access to all 35 flashcards in this deck.



